Our research focuses on the development of theoretical approaches to understand and rationally design electronic materials. Using predictive computational techniques, we apply these theoretical calculations to enable a richer understanding of the electronic and atomistic mechanisms of large material systems, especially those requiring an accurate description of electron dynamics. The field of electron dynamics is an emerging research area in chemistry and materials science because it focuses on the non-equilibrium (i.e., time-dependent) electronic behavior of material systems, which govern several chemical, material, and biological processes. A more detailed understanding of electron dynamics undergirds numerous research and technological advancements, including plasmon-mediated photocatalysis, optically induced charge separation, and light-harvesting nanomaterials. Our expertise in developing/applying new electronic structure and time-dependent dynamics techniques brings to the field a unique capability for exploring these rich and emerging areas in complex chemical/material systems:
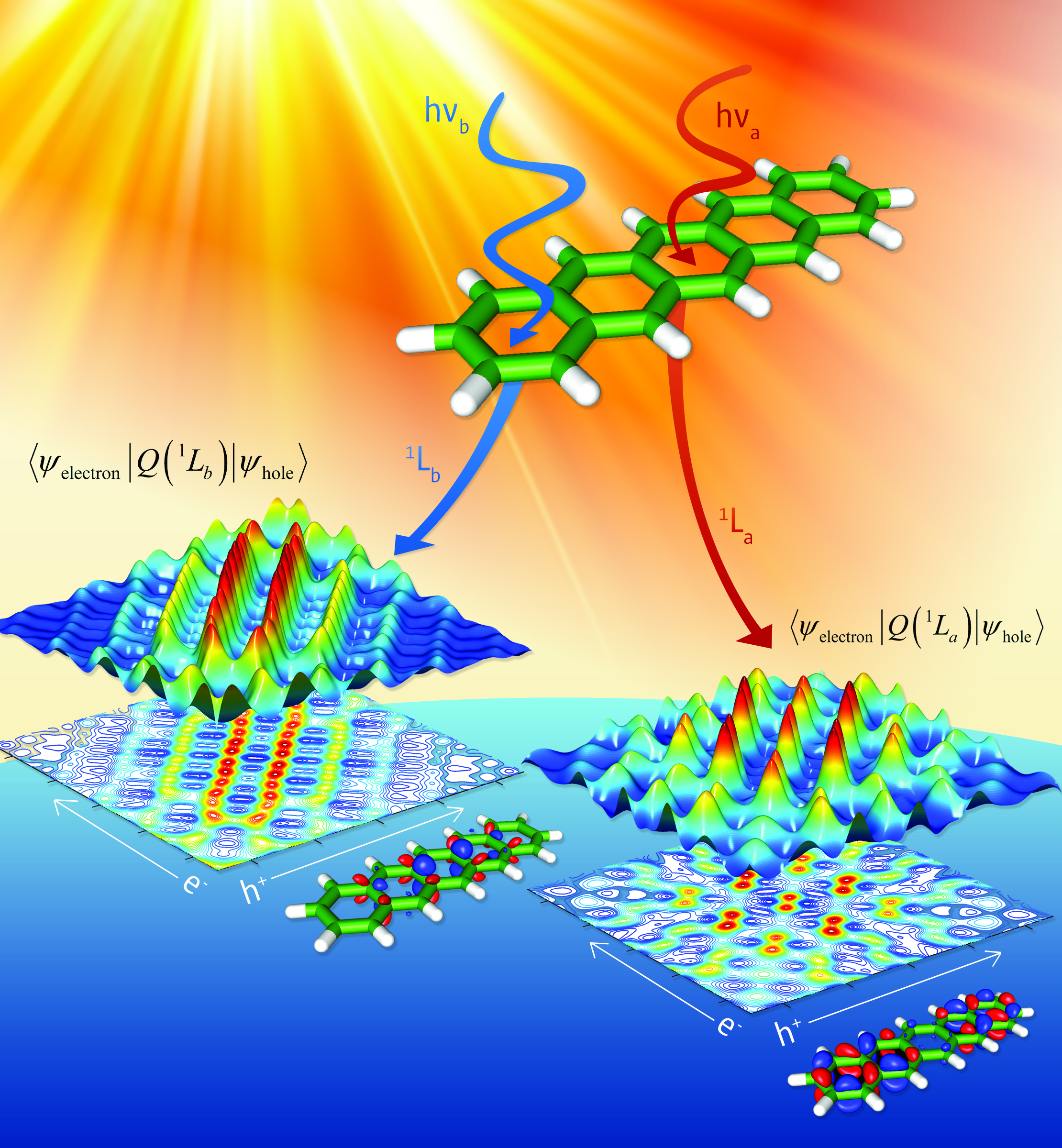 Real-Time Dynamics of Light-Harvesting Systems.The main workhorse of our research is the development and application of real-time, time-dependent density functional theory (RT-TDDFT) approaches, which are used to understand electronic-excited states and any general non-equilibrium process at the electronic/atomistic scale. While the majority of quantum calculations have traditionally been used to probe ground-state properties of chemical systems (typically as a black-box computational tool), these techniques are incapable of describing excited-state electron dynamics, which is absolutely crucial for describing any electromagnetic-induced or general non-equilibrium material process. Our RT-TDDFT techniques allow accurate and efficient calculations of light-matter interactions, optical response, photocatalysis, and photo-driven reaction dynamics in large, complex material systems.
Real-Time Dynamics of Light-Harvesting Systems.The main workhorse of our research is the development and application of real-time, time-dependent density functional theory (RT-TDDFT) approaches, which are used to understand electronic-excited states and any general non-equilibrium process at the electronic/atomistic scale. While the majority of quantum calculations have traditionally been used to probe ground-state properties of chemical systems (typically as a black-box computational tool), these techniques are incapable of describing excited-state electron dynamics, which is absolutely crucial for describing any electromagnetic-induced or general non-equilibrium material process. Our RT-TDDFT techniques allow accurate and efficient calculations of light-matter interactions, optical response, photocatalysis, and photo-driven reaction dynamics in large, complex material systems.
Further details of our work in this area are described in the following papers:
N. V. Ilawe, M. B. Oviedo, and B. M. Wong, “Real-Time Quantum Dynamics of Long-Range Electronic Excitation Transfer in Plasmonic Nanoantennas.” Journal of Chemical Theory and Computation, 129, 3442-3454 (2017). [pdf]
J. M. Rodríguez-Borbón, A. Kalantar, S. S. R. K. C. Yamijala, M. B. Oviedo, W. Najjar, and B. M. Wong, “Field Programmable Gate Arrays for Enhancing the Speed and Energy Efficiency of Quantum Dynamics Simulations.” Journal of Chemical Theory and Computation, 16, 2085-2098 (2020). [pdf]
K. Hanasaki, Z. A. Ali, M. Choi, M. Del Ben, and B. M. Wong, “Implementation of Real-Time TDDFT for Periodic Systems in the Open-Source PySCF Software Package.” Journal of Computational Chemistry, 44, 980-987 (2023). [pdf]
Q. Xu, M. Del Ben, M. S. Okyay, M. Choi, K. Z. Ibrahim, and B. M. Wong, “Velocity-Gauge Real-Time Time-Dependent Density Functional Tight-Binding for Large-Scale Condensed Matter Systems.” Journal of Chemical Theory and Computation, 19, 7989-7997 (2023). [pdf]
M. Choi, M. S. Okyay, A. P. Dieguez, M. Del Ben, K. Z. Ibrahim, and B. M. Wong, “QRCODE: Massively Parallelized Real-Time Time-Dependent Density Functional Theory for Periodic Systems.” Computer Physics Communications, 305, 109349 (2024). [pdf]
 Electronic Properties of Nanostructures and Complex Materials. Understanding and predicting the electronic properties of nanostructures and complex materials is key to tailoring experimental conditions for optimal charge and energy transfer across multiple chemical and material domains. In contrast to isolated molecules, material systems are significantly more complex since the electronic structure of extended solids/surfaces and nanostructures must be treated as a quasi-continuum. This regime lies at the crossroads of several technologically important areas, such as functional materials, self-assembled nanostructures, electrochemical systems, ferroelectric materials, and all aspects of surface science. Our expertise in quantum calculations of these material systems provides mechanistic information on energy-transfer mechanisms and dynamical processes to complement experimental efforts on nanostructures and complex materials.
Electronic Properties of Nanostructures and Complex Materials. Understanding and predicting the electronic properties of nanostructures and complex materials is key to tailoring experimental conditions for optimal charge and energy transfer across multiple chemical and material domains. In contrast to isolated molecules, material systems are significantly more complex since the electronic structure of extended solids/surfaces and nanostructures must be treated as a quasi-continuum. This regime lies at the crossroads of several technologically important areas, such as functional materials, self-assembled nanostructures, electrochemical systems, ferroelectric materials, and all aspects of surface science. Our expertise in quantum calculations of these material systems provides mechanistic information on energy-transfer mechanisms and dynamical processes to complement experimental efforts on nanostructures and complex materials.
Further details of our work in this area are described in the following papers:
C. Fu, M. B. Oviedo, Y. Zhu, A. v. W. Cresce, K. Xu, G. Li, M. E. Itkis, R. C. Haddon, M. Chi, Y. Han, B. M. Wong, and J. Guo, “Confined Lithium–Sulfur Reactions in Narrow-Diameter Carbon Nanotubes Reveal Enhanced Electrochemical Reactivity.” ACS Nano, 12, 9775–9784 (2018). [pdf]
C. Lian, Z. A. Ali, H. Kwon, and B. M. Wong, “Indirect but Efficient: Laser-Excited Electrons Can Drive Ultrafast Polarization Switching in Ferroelectric Materials.” Journal of Physical Chemistry Letters, 10, 3402-3407 (2019). [pdf]
C. Lian, Z. A. Ali, and B. M. Wong, “Charge Density Wave Hampers Exciton Condensation in 1T-TiSe2.” Physical Review B, 100, 205423 (2019). [pdf]
S. S. R. K. C. Yamijala, H. Kwon, J. Guo, and B. M. Wong, “Stability of Calcium Ion Battery Electrolytes: Predictions from Ab Initio Molecular Dynamics Simulations.” ACS Applied Materials & Interfaces, 13, 13114-13122 (2021). [pdf]
 Charge and Energy Transport in Mesoscale Materials. Mesoscale materials have garnered significant attention since they are the next step in advancing materials beyond the nanoscale regime. At mesoscale dimensions, the emergence of collective behavior naturally arises as interactions between disparate degrees of freedom (electronic, mechanical, and chemical phenomena all acting in concert) become dominant. Most importantly, quantum mechanical effects can still remain very strong in mesoscale systems, and we have developed several computational techniques and open-source software codes to treat these large, complex systems.
Charge and Energy Transport in Mesoscale Materials. Mesoscale materials have garnered significant attention since they are the next step in advancing materials beyond the nanoscale regime. At mesoscale dimensions, the emergence of collective behavior naturally arises as interactions between disparate degrees of freedom (electronic, mechanical, and chemical phenomena all acting in concert) become dominant. Most importantly, quantum mechanical effects can still remain very strong in mesoscale systems, and we have developed several computational techniques and open-source software codes to treat these large, complex systems.
Further details of our work in this area are described in the following papers:
B. M. Wong, F. Léonard, Q. Li, and G. T. Wang, “Nanoscale Effects on Heterojunction Electron Gases in GaN/AlGaN Core/Shell Nanowires.” Nano Letters, 11, 3074-3079 (2011). [pdf]
C. Chevalier and B. M. Wong, “HADOKEN: An Open-Source Software Package for Predicting Electron Confinement Effects in Various Nanowire Geometries and Configurations.” Computer Physics Communications, 274, 108299 (2022). [pdf]
Y. Chen, S. N. Sandhofer, and B. M. Wong, “SHORYUKEN: An Open-Source Software Package for Calculating Nonlocal Exchange Interactions in Nanowires.” Computer Physics Communications, 300, 109197 (2024). [pdf]
Y. Chen, M. S. Okyay, and B. M. Wong, “MISTER-T: An Open-Source Software Package for Quantum Optimal Control of Multi-Electron Systems on Arbitrary Geometries.” Computer Physics Communications, 302, 109248 (2024). [pdf]